¿Los ensamblajes genómicos reflejan siempre el tamaño del genoma?
Con la llegada de las nuevas tecnologías de secuenciación de alto rendimiento, se ha producido un crecimiento significativo del número de recursos genéticos al alcance de la comunidad científica. Con estos avances, los investigadores tenemos la necesidad de escoger entre las estrategias disponibles a la hora de analizar los datos que se generan. Seleccionar, pues, entre el abanico de aplicaciones bioinformáticas disponibles plantea nuevos retos.
Estas técnicas nos permiten llevar a cabo estudios de genómica comparada cada vez más ambiciosos, necesarios para poder entender en detalle la evolución molecular de los organismos. Uno de los factores principales a tener en cuenta a la hora de plantear un proyecto de secuenciación es el tamaño del genoma del organismo que se pretende estudiar. De hecho, en este estudio ponemos de relieve la necesidad de tener un conocimiento previo de este parámetro, no sólo para poder anticipar los costes derivados de la secuenciación, sinó también porque nos servirá para poder optimizar los análisis bioinformáticos asociados, y evaluar el rendimiento y la idoneidad de un determinado programa informático.

Figura 1. El hongo Leucagaricus gongylophorus en una colonia de hormigas cortadoras de hojas de la especie Atta colomica. Foto: Pepijn Kooij.
En este caso, hemos analizado y comparado el ensamblaje del genoma en un pequeño grupo de hongos tropicales cultivados por hormigas que pertenecen a los géneros Leucoagaricus y Cyphomyrmex y, además, hemos estimado el tamaño de su genoma utilizando citometría de flujo. Hemos comparado cinco programas de ensamblaje genómico y hemos observado que el tamaño del genoma obtenido en cada caso depende directamente del programa utilizado, y que además impacta en la predicción del número de genes que prevé cada programa. Debemos tener en cuenta que los análisis bioinformáticos pueden verse influenciados por el grado de heterozigosidad del genoma, la poliploidía o el contenido de ADN repetitivo. Curiosamente, el análisis de los datos publicados en este caso indica que el tamaño del genoma obtenido a través del análisis de secuencias de ADN es de 2,3 a 3 veces mayor que cuando utilizamos la citometría de flujo.
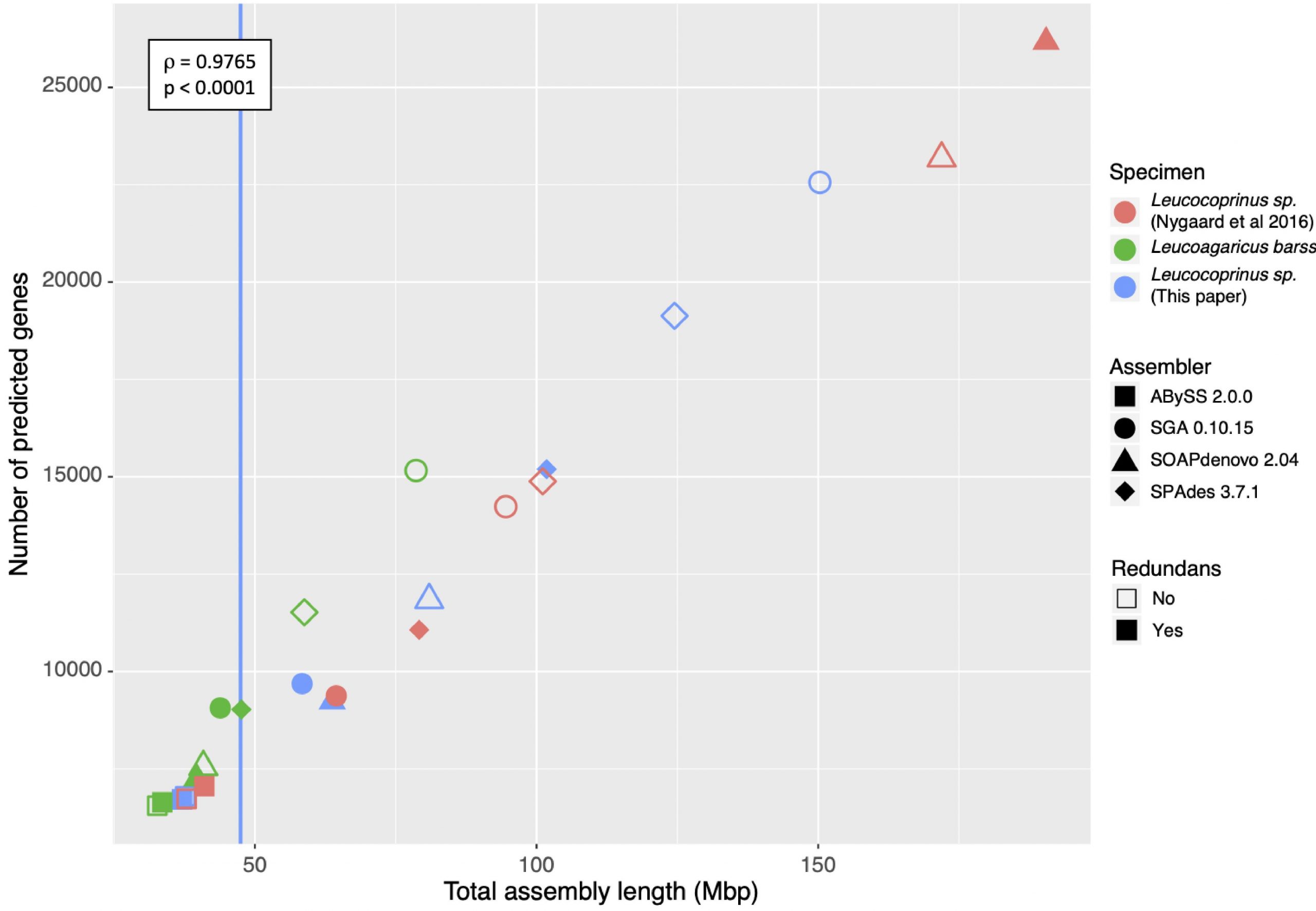
Figura 2. Correlación entre el tamaño del ensamblaje y la predicción del número de genes basada en los análisis con cuatro programas bioinformáticos diferentes.
En base a estos resultados recomendamos calibrar y refinar los análisis integrando datos de citometría de flujo como complemento a los análisis bioinformáticos, para poder tener una visión más precisa sobre el comportamiento de cada programa y establecer predicciones sobre el tamaño del genoma con mayor robustez.
Referencia:
Kooij, P. W., Pellicer, J.. (2020) Genome size versus genome assemblies: are the genomes truly expanded in polyploid fungal symbionts? Genome Biology and Evolution. https://doi.org/10.1093/gbe/evaa217